
Research Article | DOI: https://doi.org/10.31579/2693-4779/040
Retired in Department of hematology, Iran University of Medical Sciences, Tehran, Iran.
*Corresponding Author: Ahmad Reza Rahnemoon, Retired in Department of hematology, Iran University of Medical Sciences, Tehran, Iran.
Citation: Ahmad Reza Rahnemoon. (2021) The genetic abnormalities investigation of leukemia in past few decades. Clinical Research and Clinical Trials. 3(3); DOI:10.31579/2693-4779/040
Copyright: © 2021 Ahmad Reza Rahnemoon. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 29 April 2021 | Accepted: 04 May 2021 | Published: 06 May 2021
Keywords: leukemia; cytogenetic; molecular analysis; lymphoid stem cells; myeloid progenitor cells
Cytogenetic and molecular analysis of each patient’s leukemia cells has become an essential component of diagnosis prior to treatment. It has furthered our understanding of leukemogenesis at a molecular level. Specific and well-characterized recurring chromosomal abnormalities facilitate diagnosis, confirm subtype classification, and have major prognostic value for treatment planning. Conventional chromosome analysis is a basic way for diagnosis and treatment. In addition in this way evaluation of disease progression is important and so it is the only method that can identify the presence of clonal evolution, particularly in accelerated and relapse phase in the disease. Also conventional cytogenetic can detect chromosomal abnormality associated with its advanced phase. Anyhow, the value of translocation rates in interphase and metaphase nuclei in monitoring leukemia is at the time of diagnosis and after treatment additionally. Genomic profiling transformed our understanding of the genetic basis of leukemia particularly in acute leukemia, which is a malignant clonal proliferation in lymphoid stem cells or myeloid progenitor cells.
As we know, leukemia is the consequence of step-wise genetic alterations that confer both proliferative and survival advantage, as well as self-renewal capacity to the malignant cells. Leukemia stem cells (LSCs) possess several key properties of normal cells including self -renewal, unlimited proliferative potential, infrequent or slow replication. LSCs infiltrate the bone marrow and interfere with the normal HSC microenvironment hemostasis. Thus, the major difference between leukemia growth and normal tissue renewal is that whereas normal transit amplifying cells usually differentiate and die at various levels of differentiation, the leukemia transit-amplifying cells can go to differentiate abanormally and instead, accumulate, resulting in leukemia growth(1-.2,7-12)We know cytogenetic and molecular testing in leukemia is integral to diagnosis and minimal residual disease (MRD) monitoring as well. For fusion genes studies FISH is a more sensitive test in the advantage of routinely interrogating 50 to 200 metaphase or interphase cells .However, one of the most sensitive test is RT-PCR in molecular fusion gene study and MRD assay as well[1,28,39].
I-Methods:
The total chromosomal number, followed by the sex chromosomes, numerical and structural aberrations according to the International System for Human Cytogenetic Nomenclature. Viz, at least two cells with the same structural rearrangement, like translocations, inversions, deletions, gain or loss of chromosome can be considered as an abnormal clone which represents the malignant cells karyotype in any cancerous. These cytogenetic abnormalities are acquired somatic (rather than germ line) mutations that frequently result from translocations of chromosomal DNA, resulting in new (abnormal) protein products from the resultant fusion genes. It is assumed that the protein products from these fusion genes are responsible for the cellular dis regulation that leads to the malignant state. Thus, we can say deletions or loss of DNA may eliminate genes that have tumor suppressor functions or gains of additional chromosomes may lead to gene dosage effects that provide transformed cells with survival advantages.
Conventional cytogenetic analysis, requires dividing cells, is technically difficult, and can be time-consuming due to the presence of multiple abnormal cell lines and complex chromosomal banding patterns. In fact, alternative diagnostic methods have been sought, including flourescence in situ hybridization (FISH), in which labeled probes are hybridized to either metaphase chromosomes or interphase nuclei and then detected with fluorochromes. This method of analysis is more rapid and in some cases more sensitive, than conventional cytogenetic analysis. Additionally, FISH can be used to study differentiated or non-dividing cells. Namely, FISH with the appropriate probes can use to detect early relapse, residual disease and/ or in assessment of therapeutic regimens efficacy [38, 50].
As we know, the first aid in order to identify most of the specific chromosomal aberrations appearing in bone marrow peripheral blood cells in patients with ALL is classical cytogenetics. It allows the determination of the clinical significance of these abnormalities as well. However, in case of t (12:21) (p12; q22), it is useless because of submicroscopic nature of this translocation and hence, the classical cytogenetics is not a method sensitive enough to identify it. Therefore, being based only on this technique, it affirms the normal karyotype that has very severe consequences in subsequent treatment decisions. These issues are discussed in the following part.
The more sensitive and credible technique is fluorescent in situ hybridization (FISH) that allows the analysis not only of the cells arrested at the metaphase, but in interphase nuclei as well[41]. With the usage of dual-color whole chromosomes painting probes for 12 and 21 chromosomes or specific probes for TEL and AML1 loci, the identification of t(12;21)(p12;q22) is effective. The changes described above: Del (12) (p12) and additional copies of AML1 gene are also detectable by FISH [27, 38, 47-50].
The expression outcomes are consistent with FISH results, but there are discrepancies. Some patients are positive using RT-PCR, but negative with regard to FISH. Conversely, the lack of expression using RT-PCR, but there is the presence of fusion signals. The atypical breakpoint on TEL gene and also the methods sensitivity may be the explanation of these incompatibilities [42].
The methods presented above are useful not only to diagnose the patients at the beginning of illness, but also to control them during therapy, remission and relapse. The detection of chromosomal markers by means of classical cytogenetics at diagnosis is not sufficient for the classification of patients in different risk groups. Thus, the classical techniques should be strictly connected with molecular, very sensitive analysis such as: FISH or RT-PCR that allow to identify one leukemic cell among 104 or 105 normal ones.
Nowadays, genetic and molecular analyses have been widely used in practice to support the clinical criteria which have been used. ALL in children is a heterogeneous disease with different subtypes based on their molecular features. This condition will influence the treatment response and risk for relapse. Thus, a good early stratification is needed to achieve outcome. As it is not a single disease, it may be distinguished from other malignant lymphoid disorders by the immunophenotype of the cells [1, 28, 47-50].
II- Genetic abnormalities investigations:

It can be with multiple trisomies and between 50 and 66 chromosomes (normal=46) is referred to as hyper-diploid. This usually occurs in children and implies favorable prognosis. Trisomies of chromosome 4, 10, and 17 are considered markers for low- risk disease in pediatric leukemia protocols (table 1). I have some points as follows: 1) as we know karyotype can be abnormal in ALL and this abnormality including numerical aberration and structural aberration. In structural aberration, if we had some chromosomal involved or multiple involved, we can say, it is a complex karyotype that generally we see in accelerated phase or in relapse with poor prognosis. So in last decades, at response of many questions were not accurate and not clear [3-8, 10-17, 29, 47]. Told all, in table 1 we have different chromosomes in involved cell lines. The question is, why is different in involved chromosomes at ALL [29, 47, 50]? Suppose we have ALL-L1, hence why so different in chromosomal abnormalities response in one of the subtype of ALL? In the table, what about a minor clone? Look at the table again, you see 4,6,8,10,14, 17, 19 and 22 were chromosomal involved. What does it mean exactly? In other words, a lot much of human chromosomes were involved in a kind of ALL patients. Why? Also we can say
These chromosomal changes or molecular abnormalities may accidental or fortuitously do so. Thus, the molecular genetics abnormalities is debated in the following of my discussion.
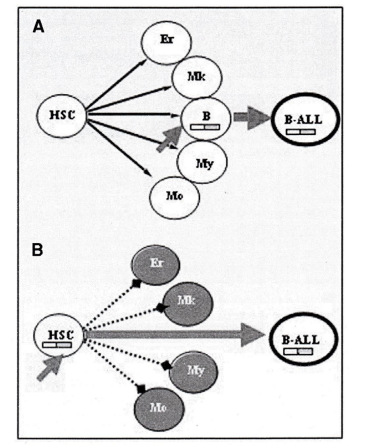
Figure 1: A fusion gene model and chromosomal abnormalities in the cell lineage specification, seeing that cell abnormalities forces expression of these fusion genes in lineage choice decesions, which imposed in the lineage outcome [46].
Anyhow, I have some problems as follows:
As we know lymphoid malignancies are characterized by recurrent chromosomal aberrations that lead to the formation of fusion genes and the subsequent expression of chimeric proteins with unique properties. The best example concerning the necessity of secondary events is given by leukemias with ETV6?RUNX1( E/R) fusion gene. Generally, t(12;21) positive patients show frequent deletion of the normal ETV6 gene, suggesting that the fusion gene is an initial event conferring predisposition to leukemia by the deletion of the gene in 12p as a promoter event (Fig. 1).
Genomic sequence analysis of E/R and other fusion genes suggests that the predominant mechanism of chromosome translocation is double-stranded DNA breakage followed by normal but error-prone repair by non-homologous recombination [27, 30]. However, this begs the question of what causes the initial DNA damage. For MLL gene fusions, there is experimental genetic and epidemiologic [38]. Evidence that transplacental chemical carcinogenesis may be involved. However, what might be responsible for the relatively high frequency (~1%) of E/R fusion generation in normal fetal hematopoiesis? Presumably, nonfunctional fusions (in non-stem cells) occur at an even higher rate. One possibility is that these are ‘normal’ developmental errors of DNA maintenance reflecting the complexity of embryo and fetal tissue engineering in which cell death, DNA damage and oxidative stress are ubiquitous [30]. The initiation of other pediatric cancers has similarly been ascribed to developmental accidents [39]. Even if E/R fusion is in effect, a spontaneous error, the risk of this event occurring may be modified by other factors. There is dietary and genetic evidence that folate has an impact on the risk of infant and childhood leukemia, and this could well be operative during pregnancy in utero, influencing the likelihood of chromosomal breaks [35-37].
Whatever the mechanisms governing chromosomal changes in utero, it is evident that for most cases of childhood leukemia, the crucial etiologic bottleneck is the postnatal ‘promotional’ events or exposures that precipitate the secondary genetic changes, including TEL deletion. Uncovering the nature of these exposures is the objective of major case/control epidemiologic studies in Europe and the US. Although no consensus view has yet emerged, there is now a body of evidence implicating infections in childhood ALL [48-49].
In spite of the E/R fusion protein induces a silent pre-leukemic clone which requires genetic hits for the transition to leukemia as well. In positive E/R, the early genetic lesion is followed by a number of driver that it means in copy number alterations (CNAs) including loss of ETV6 and also other alterations in regulating of normal B-cell differentiation which acquired a dynamic clonal architecture. E/R fusion drives not only the process of leukemic transformation, but also the maintenance and leukemic cells generation. At last, ALL relapse arises from the emergence of a minor sub-clone and additional distinct genetic alterations from predominant clone in diagnosis. So, in this case, the patients should respond differently to the anti-leukemic therapy, which ultimately can try to improve outcome for high risk ALL patients. Also, the CNAs in relapse specifically, not only for the relapse development, but also to drug resistance too [27-28 48, 50]
So I have some points: 1) Studies with E/R knock in mice showed that the expression of the fusion gene is not sufficient for the invivo induction of ALL. 2) Why the translocation t (12; 21) or E/R fusion gene can be detected in healthy individuals? I may say, why can be detected and what about the result? Is it accepted that we say, in this case, likely to be indicative of transient genomic instability? Or is it better, those are complex that we do not understand? In fact, can these solve? 3) Some researchers believe that leukemia associated fusion genes before birth. They say, in twins /with concordant E/R positive leukemia, the development of ALL has been found to occur at different times, and postnatal latency can be variable and occasionally protracted; or somebody say ,E/R translocation occurs in utero, followed by pre-leukemic evolution as a result of further genomic structural variation. In other words, pre-leukemic clones carrying E/R oncogenic lesions which found in neonatal cord blood, but a little carriers developed to B-cell leukemia and we know that the expression of E/R oncogene is restricted to the stem cell compartment in a mouse model as well. In fact, E/R fusion protein induces a silent pre leukemia clone which requires additional genetic abnormalities in changing to leukemia as well [3-7, 14-17].
Over all, these findings are accordance with the results of conventional cytogenetic and FISH analysis and provide a further example of molecular mechanism for the paradigm of E/R fusion accompanied by deletion of the residual TEL allele. It also pointed fusion gene as a favorable prognosis in the therapeutic protocol and early detection of the disease. The high rate of early B-lineage ALL patients (Fig.2) carrying this translocation might consider as a presumption that expression of the fusion product hampers the in vitro growth and establishment in the culture of such leukemic cells.
Firstly, this rearrangement and the transcription factor AF4 which results from t(4;11)(q21;q23) were identified from normal children and fetuses as well as fetal liver samples. Then some researchers , the rearrangement detected in healthy individuals. Sequence analysis of individual providing evidence that these rearrangements are not restricted to malignant cells and that they may also be present in a subset of normal hematopoietic cells. Secondly, abnormalities of the MLL gene at chromosome region 11q23 are seen in the 5% to7% of cases of ALL, as well as in AML. We can say , translocation t(11;19)(q23;p13.3) is the next most frequent MLL abnormality in ALL but also occurs in AML(monocytic)What does it mean? We have the fusion gene abnormality in two different lineages including myeloid series and the other lymphoid series. Is this fusion gene a specific abnormality or nonspecific abberation? What about the production of the fusion gene in two different lineages? Are these the same?[9-15,17-18]Therefore, we must have a better understanding about the interaction of HSCs with stromal cells and other important agents like genetic abnormalities such as cytogenetic and molecular genetic which can provide therapeutic targets for manipulation to modulate HSCs and their progeny in the diseases, in addition, the rearrangement has been reported in normal persons too.
It was as the first chromosomal abnormality shown with a specific malignancy in humans. In this regard, murine models have demonstrated that disruption of the hematopoietic microenvironment can initiate myelo-proliferative disease and even leukemia. Also in murine models, activation of the normal from HSC niche improves recovery from radiation and chemotherapeutic injury and suppresses chronic myeloid leukemia( CML) progression, impairing leukemic stem cell maintenance in the syngeneic model. In hematologic malignancies, clonal neoplastic cells alter the hematopoietic microenvironment so that it becomes supportive of LSCs and becomes less supportive of normal HSCs, ultimately leading to decreased normal hematopoiesis. In CML , sustained by a range of biological characteristics that enable their long-term survival, and accumulation of myeloid cells that differentiate in normal and abnormal clones, which can change to acute lymphoblastic leukemia(ALL) in accelerated phase possibly. Furthermore, leukemia induced decrease in CXCL12 expression results in reduced retention of LSCs in CML bone marrow. Moreover, leukemia induced abnormalities in cytokine in CML bone marrow result in selective suppression of normal stem cell growth and enhanced growth of LSC [3-4,33-36].
The Ph’ chromosome t(9;22) with a production of a fusion BCR/ABL occurs in about 30%of cases of adult(20?tected at karyotyping) and 6% of cases of childhood ALL .The t(9;22) is the most frequent adult ALL translocation and is associated with poor prognosis. Two types are found: that identical to CML, involving the bcr region of BCR with resultant p210 kD fusion protein, occurring in half of adult cases of Ph’ positive ALL; and another p190 kD protein that occurs in most childhood ph’ positive cases. Regarding , I have some points: 1) What is the difference between CML and ALL? Are these the same? Because of the proteins of 210 and 190 can be seen in ALL and also in CML? How is it possible? Also, I must say, we have two different lineages completely that producing two different kind of cells So, is this mechanism right? 2) Some researchers stated that BCR/ABL fusion gene detected in normal individuals. What does it mean? And in other words, what is the exact role of this fused gene in the individuals?[19-26,31-32]

Figure 2: The role of genetic alterations in the B-ALL pathogenesis. These lesions with or without the secondary genetic alterations disrupt lymphoid lineage and go to a maturation arrest, that result in resistance to therapy and promote to disease relapse probably [45].
The cytogenetic analysis are completed by expression study of E/R on der (21) and TEL/AML1 on der (12) by means of RT-PCR that makes possible the finding of mRNA. The presence of transcript on der (21) is observed more frequently than AML1/TEL chimeric transcript, showing that the protein encoded by E/R plays the crucial role in leukemogenesis process [40, 3].
However, hypothesis that the E/R protein generates and maintains pre leukemic clones and that conversion to overt disease requires secondary genetic changes, possibly in the context of abnormal immune responses [43, 44], is justified by our finding of age range of patients by positive fusion gene (Fig.2). These results are an appropriate issue to attempt of achieving a good early stratification for better outcomes and suggesting a combination of cytogenetic and molecular profiling to permit more accurate assessment of the disease prognosis and improving molecular sub-classification to allowing stratified management. In addition, in order to indication of some studies the t (12; 21) translocation in children occurs before birth, in embryo phase, it is suggestible to use molecular detection test in the period. The initiating lesion happen in utero and after birth go to the pre-leukemic state. Infection can be as a unregulated immune response in a little of pre-leukemic children and after secondary mutations, the children develop to ALL, to lead a clonal expansion ultimately. But the main questions are as follows: why most pre-leukemic children remain healthy? Why some rearrangements like KMT2A are not sufficient for developing ALL? Why some rearrangements like BCR/ABL, E/R can be indicated in normal persons without apparent oncogenic consequences? It is not completely clear that what is the exact role of these fusion genes in this group? Is there a transient genomic instability as a fact in the healthy individuals? [1, 35, 39, 48-49]
In the end, conventional cytogenetic, FISH technologies and RT-PCR have a significant role in detecting the abnormalities, but microarray and next generation sequencing(NGS) could prove useful in the duration of disease from diagnosis till treatment because they must satisfy a major role in the diagnosis and fit a new look in management of hematological disorders as well.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
